The rise of small molecule drugs discovered through artificial intelligence is a notable trend where computer science and life science converge. Fragment-based drug discovery (FBDD) has emerged as a new approach in the process of finding new compounds. The Generative Pre-trained Transformers (GPT) model has demonstrated its extraordinary abilities in various fields, thanks to its pre-training and representation learning of basic language units. Similar to how natural language operates, molecular encoding as a form of chemical language, requires careful fragmentation according to specific chemical principles to achieve precise encoding.
Significance of Fragmentation
FBDD has become an effective tool for discovering new drugs, gradually emerging as a significant alternative to the traditional approach of identifying lead compounds through High Throughput Screening (HTS). Unlike HTS, FBDD can identify smaller chemical components. By utilizing the concept of fragmentation, FBDD can obtain molecular fragments that interact with different fragments of the biological target molecule. The extraction and analysis of fragment-target interactions are key focuses in FBDD research. Compared with HTS, FBDD offers distinct advantages. Its main advantage lies in its ability to identify hits in a wide chemical space while screening only a limited number of compounds. FBDD provides better opportunities for generating lead compounds with drug properties.
The emergence of GPT has sparked widespread discussion on its impact on drug development. Traditional natural language processing (NLP) research mainly relies on syntax and rules meticulously designed by experts. However, in recent years, the introduction of Transformers and attention mechanisms has enabled models to master various relationships between tokens in the input sequence. This paradigm shift has drastically changed the field of sequence data processing and made key breakthroughs in understanding natural language text. The chemical structure of a molecule can be represented using a linear encoding similar to the representation of natural language text. Similar to sentence segmentation in NLP, the description of molecular fragments is also analogous to this concept. In the wider chemical space, molecular fragments made up of atoms and bonds can also construct more accurate representations, providing greater flexibility for the molecular representation process.
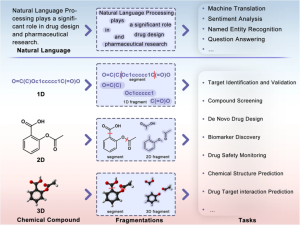
Molecular fragmentation technologies benefit downstream tasks.1
However, the current challenge lies in how to select molecular fragments with complete chemical activity, maintain their integrity, and avoid excessive expansion. The traditional approach often relies on fragment libraries maintained by many companies and drug research teams for a long time. However, the high costs of fragment acquisition, limited types of fragments, and uneven distribution of fragments limit its widespread use in conventional drug discovery. In view of the significant trend of artificial intelligence molecular fragmentation and the limitations of the current use of fragment libraries, the use of large-scale and non-professional-dependent fragmentation methods has become an important direction for future research.
Method of Fragmentation
Molecular fragmentation is the process of breaking down large molecular compounds into smaller fragments, which may consist of individual functional groups or complex fragments with specific structural characteristics. Molecular fragments find wide applications in computational chemistry, drug design, and chemical informatics. This article focuses on systematically classifying and organizing methods related to molecular fragments, including the mode of molecular fragmentation, whether specific structures (such as cyclic structures or double bonds) are destroyed in molecular fragmentation, preserved fragment information, and the insertion of preset fragment libraries, etc.
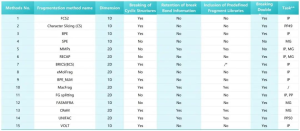
Summary of Fragmentation Methods.1
Applications of Fragmentation Techniques
Building high-quality molecular fragment libraries is crucial for FBDD. In the process of drug development, traditional computer-aided drug design methods often need to obtain complete drug molecular information for computer simulation. By encoding input data and training, a reliable functional application model can be obtained. However, FBDD is a completely different drug discovery method from traditional high-throughput screening methods. The advantage of FBDD is its high sensitivity to small molecular fragments and a relatively small compound library, making it more likely to discover drug candidate molecules that may be overlooked by other screening methods. However, how to choose a suitable molecular fragmentation method and better exploit the advantages of FBDD in practical applications remains one of the main problems that plague researchers.
Selection of Fragmented Molecules
When the purpose of molecular fragmentation is to promote molecular expression, the selection of fragments after fragmentation becomes particularly important. Screening the fragment library generated by fragmentation is an important step in obtaining high-quality fragments. Although this selection may result in some loss of information, the selected molecular fragments can effectively capture the inherent chemical properties of the molecule. Under a FBDD context, fewer heavy atoms are used to screen compounds compared to traditional high-throughput screening devices. Hit identification methods must adapt to smaller fragment sizes, necessitating sensitive biophysical techniques or higher concentrations for biochemical assays. Theoretically, a fragment library can cover a larger proportion of chemical space compared to a high-throughput screening (HTS) library, providing more confidence for researchers in building molecules from the fragment library. When constructing a filtered molecular fragment library, the first consideration is the number of compounds in the fragment library, which is largely driven by the detection technology used for fragment screening. High-throughput technologies, such as high content screening (HCS), typically utilize biochemical assays and are usually less sensitive compared to low-throughput biophysical techniques. Generally, less sensitive techniques require more effective fragments which may involve more complex compound fragments, signifying the need for a larger fragment library. At the same time, with the increase in ligand complexity, the possibility of any fragment being hit exponentially decreases. Therefore, it is necessary to ensure that the library: 1. Samples the relevant chemical space by merging key pharmacophoric groups that drive fragment binding. 2. Combines appropriate distribution and balance with fragments of sufficient complexity and different shapes. 3. Covers a comprehensive range of accessible fragment diversity to effectively optimize fragment hits in lead compounds. Avoid clustering associated with known and highly reactive, solution aggregation or persistent false positive data.
Small fragments often generate hits with low affinity and specificity, but these fragments can extract features related to protein interaction. Low specificity has two consequences: firstly, fragments can bind to multiple proteins; secondly, fragments can bind to a single protein in multiple ways. In the first sense, low specificity can increase the hit rate of fragment screening because specificity can be introduced later during fragment optimization. The ability of fragments to bind to proteins in various ways may hinder optimization strategies, as these strategies assume a consistent binding pose when establishing structure-activity relationships. However, fragments with multiple binding modes are still valuable for druggability studies. They can be integrated into a comprehensive program to experimentally and computationally probe proteins with very small compounds that bind to proteins with a small number of non-hydrogen atoms and form clusters consistent with known inhibitor locations. In addition, the extension and relative position of these clusters bring important information about protein druggability.
Summary
This article has highlighted new directions or outstanding issues for future work. The first direction is to expand methods of molecular fragmentation. Energy-based cleavage methods can avoid cutting high-energy chemical bonds that are difficult to break, thereby preserving functional group fragments in chemical reactions. The second direction is to preserve fragment information. After molecular fragmentation, in addition to retaining structural information, it should also include the position, energy, relevance, etc. of the fragment in the original molecule. The final direction is to evaluate the quality of molecular fragments. In addition to relying on downstream task performance evaluation, a system needs to be established to directly evaluate the quality of molecular fragments from the fragments themselves. This ensures the fairness of molecular fragment quality assessment.
Reference
- Jinsong, Shao, et al. “Molecular fragmentation as a crucial step in the AI-based drug development pathway.” Communications Chemistry7.1 (2024): 20.