The Enigma of Artificial Intelligence (AI)
AI is evolving rapidly, yet for many, the robust models remain a ‘black box.’ Understanding the inner workings of these models and the process of their conclusions is out of reach. However, a significant breakthrough was recently achieved by Professor Jürgen Bajorath, a cheminformatics expert at the University of Bonn, and his team. They developed a technique that reveals the operating mechanisms of certain AI systems used in drug research. Their findings indicate that these AI models rely primarily on recalling existing data, rather than learning specific chemical interactions, to predict drug efficacy. In other words, AI’s predictions are assembled from memory, and machine learning is not actually occurring. These findings were recently published in the ‘Nature Machine Intelligence‘ journal.

The Role of AI in Medical Research
In the medical field, researchers are frantically seeking effective active substances to fight diseases, but finding effective compounds is like finding a needle in a haystack. Consequently, researchers initially use AI models to predict which molecules can best dock with their target proteins and bind firmly. After this initial AI-led identification, these potential drugs are explored in greater detail through experimental research.
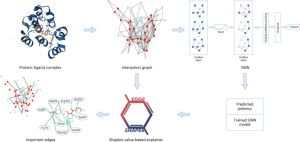
AI employs advanced techniques such as Graph Neural Networks (GNN) in the domain of drug discovery research. GNNs are applicable in predicting the binding strength of a molecule to a target protein. They use graphs, consisting of nodes representing objects and edges representing relationships between nodes. In the graph representation of protein-ligand complexes, edges connect protein or ligand nodes, indicating molecular structure or interactions between proteins and ligands. The GNN models use protein-ligand interaction graphs extracted from X-ray structures to predict ligand affinity. For many, these GNN models are like a black box, with their prediction process shrouded in mystery.
Inside the Workings of AI
Researchers from the University of Bonn’s cheminformatics collaborated with colleagues from the Sapienza University of Rome to analyze whether Graph Neural Networks truly learn the interactions between proteins and ligands. They employed their specially developed ‘EdgeSHAPer’ method to examine six different GNN architectures. EdgeSHAPer can determine whether GNNs learn the most important interactions between compounds and proteins, or if they make predictions via other methods.
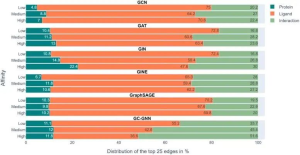
The scientists trained six GNNs using graphs extracted from protein-ligand complex structures, knowing the compounds’ modes of action and their binding strength to target proteins. The trained GNNs were then tested on other complexes, and EdgeSHAPer was used to analyze how GNNs produced predictions. According to the research team’s analysis, most GNNs did not act as expected. The majority only learned some interactions between proteins and drugs, focusing mostly on ligands.
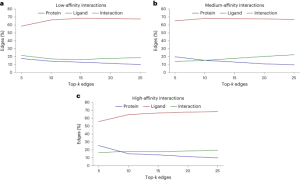
This finding leads to the ‘Clever Hans effect,’ where just as a seemingly math savvy horse actually inferred the desired outcome from subtle facial expressions and gestures of others, the GNNs’ alleged ‘learning ability’ may not hold water. Their predictions are largely overestimated as equivalent quality predictions can be achieved using simpler methods and chemical knowledge. That said, the study also discovered a phenomenon: when the potency of the test compounds improved, the model tended to learn more interactions. Perhaps by modifying representations and training techniques, these GNNs can be further improved. However, any assumptions that physical quantities can be learned based on molecular graphs should be treated with caution.
In conclusion, as AI advancements continue to revolutionize diverse sectors, including medicine, the mechanisms behind these sophisticated models remain elusive. An in-depth understanding of how these models operate is crucial for optimizing their use and developing more effective solutions in the future.