As operators of Creative Biolabs’ AI Station, we’ve watched artificial intelligence move natural-product (NP) drug discovery from scattered experiments to data-driven pipelines. A recent perspective captures this shift with unusual clarity: AI is no longer peripheral to drug discovery—it’s embedded in how we search, score, and synthesize leads, including those sourced from nature’s vast chemical space. The article maps current applications, flags persistent bottlenecks, and outlines credible next steps. Below is a practitioner’s summary and takeaways for teams building NP-centric programs with AI at the core.
Why Natural Products Plus AI Matter in Drug Discovery—Right Now
Natural products have long been a rich source of bioactive scaffolds and first-in-class mechanisms. What’s changed is the digital substrate: NP databases and multimodal datasets have expanded, enabling robust AI models to mine, rank, and generate chemistry with higher signal and less guesswork. In short, the field is shifting from manual dereplication and trial-and-error screening to model-guided discovery and design.
This transformation depends on data architecture as much as on algorithms. NP-focused AI programs benefit from standardized taxonomies, well-linked knowledge graphs, and curated property/activity metadata—foundational elements that make models generalizable rather than brittle or project-specific.
What AI is Already Doing Well in Natural Product Drug Discovery
1) Prioritizing Bioactive Chemical Space
AI-enabled cheminformatics now routinely navigates vast NP libraries, ranks analogs, and visualizes where “privileged” regions of bioactivity may lie. Examples include deep-learning and ML screens that uncovered microtubule-modulating NPs, JNK1 inhibitors, and potent anti-osteoporosis leads—each validated with prospective assays. These studies demonstrate that well-trained models can meaningfully enrich hit rates against real biological targets.
On the antimicrobial front, AI has produced remarkable results—from early “halicin-style” breakthroughs to deep-learning campaigns targeting priority pathogens such as Acinetobacter baumannii. These examples highlight AI’s ability to reveal non-obvious chemical matter beyond classical antibiotic families—an area where NPs excel.
2) Learning From Biosynthetic Gene Clusters and Omics
NP drug discovery often starts upstream of structure—within genomes and metabolomes. Machine learning on biosynthetic gene clusters (BGCs) can predict likely bioactivities, helping teams decide which strains or pathways to prioritize before committing scarce wet-lab resources. Combining this with metabolomics-guided exploration further tightens the discovery loop, guiding isolation toward chemistry that’s both novel and relevant.
3) Scoring “Natural-Product Likeness” and Filtering Feasibility
Before synthesis or purchase, models such as NP-Scout assess “natural-product-likeness,” enabling medicinal chemists to prioritize candidates that preserve NP-like topology and stereochemical richness—features often correlated with biological performance. These filters sit alongside ADMET, selectivity, and off-target predictions to shape tractable shortlists for experimental validation.
4) Generative Design Tuned to NP Chemistry
Generative transformers, VAEs, and RL frameworks have evolved from theoretical concepts into practical tools for designing “NP-inspired” scaffolds and pseudo-natural products. These models can generate molecules that retain NP-like features while simplifying synthesis—a strategy validated by numerous case studies transitioning from complex NP prototypes to bioactive, synthesizable analogs.
5) Planning How to Make It (Before You Try)
Modern retrosynthesis planners now play an essential role in AI-assisted NP drug discovery workflows. When a model proposes an NP-like candidate, synthesis planners evaluate routeability and building-block availability, reducing iteration cycles and directing chemists toward the most feasible ideas.
6) Knowledge Graphs and Multimodal Fusion
NP discovery benefits from integrating structure, bioactivity, biosynthetic, and spectral data into connected knowledge graphs. This multimodal integration supports more informed in silico target fishing, drug repurposing, and side-effect modeling, providing deeper biological context and enhancing model interpretability.
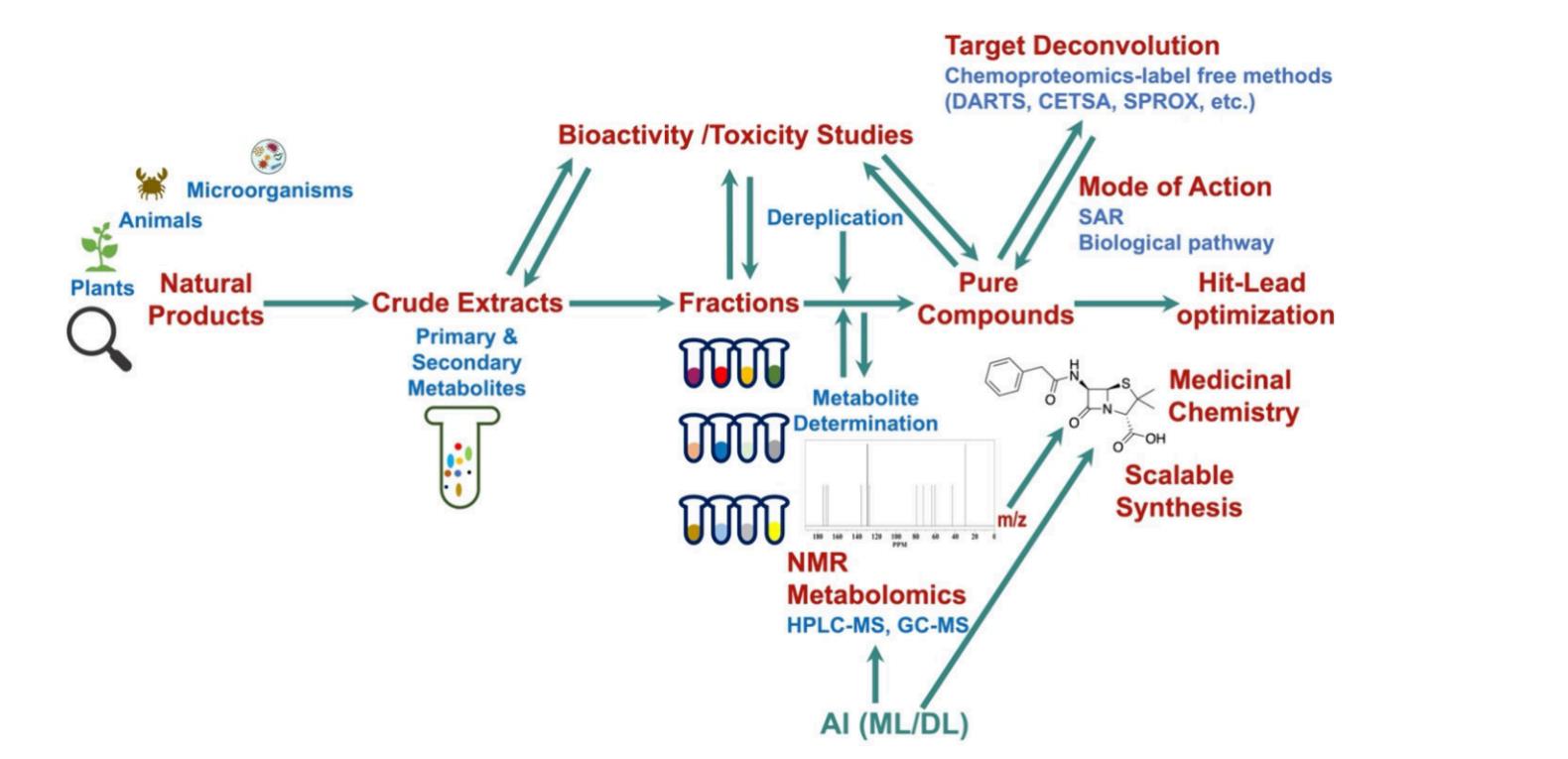
Fig.1 Overview of the NP-inspired drug discovery strategy.1
Repurposing With Graphs: Fast Paths to Proof-of-Relevance
Drug repurposing is particularly valuable in NP research, where pharmacology may be broad but under-characterized. Heterogeneous graphs, cross-network embeddings, and similarity-network fusion have been employed to infer drug–disease and drug–target links, revealing repositioning opportunities or polypharmacology worth exploring. Practically, this accelerates the transition from anecdotal signals to ranked hypotheses for phenotypic rescue or pathway modulation—followed by focused experimental validation.
The Stubborn Challenges—and Credible Fixes
1) Data Scarcity and Heterogeneity
Many NP datasets remain sparse, inconsistent, or trapped in non-standardized formats. Treating data engineering as a first-class priority—harmonizing identifiers, normalizing bioassay contexts, and adopting computable taxonomies—can dramatically improve learnability and model transferability. Transfer learning, self-supervised models, and few-shot learning have shown particular promise in low-data NP environments.
2) Dereplication and Isolation Bottlenecks
AI can rank unique chemistry, but laboratories still struggle with dereplication, scale-up, and micro-scale compound isolation. These issues are ideal targets for predictive analytics—using model-guided fractionation and LC-MS/MS prioritization to direct resources toward the most novel and promising fractions.
3) Explainability for Decision Support
As NP projects increasingly rely on GNNs and transformer-based QSAR, interpretability becomes essential. Explainable AI (XAI) tools for feature attribution and uncertainty estimation help scientists trust, understand, and act upon model predictions, enabling more informed structure–activity relationship (SAR) decisions.
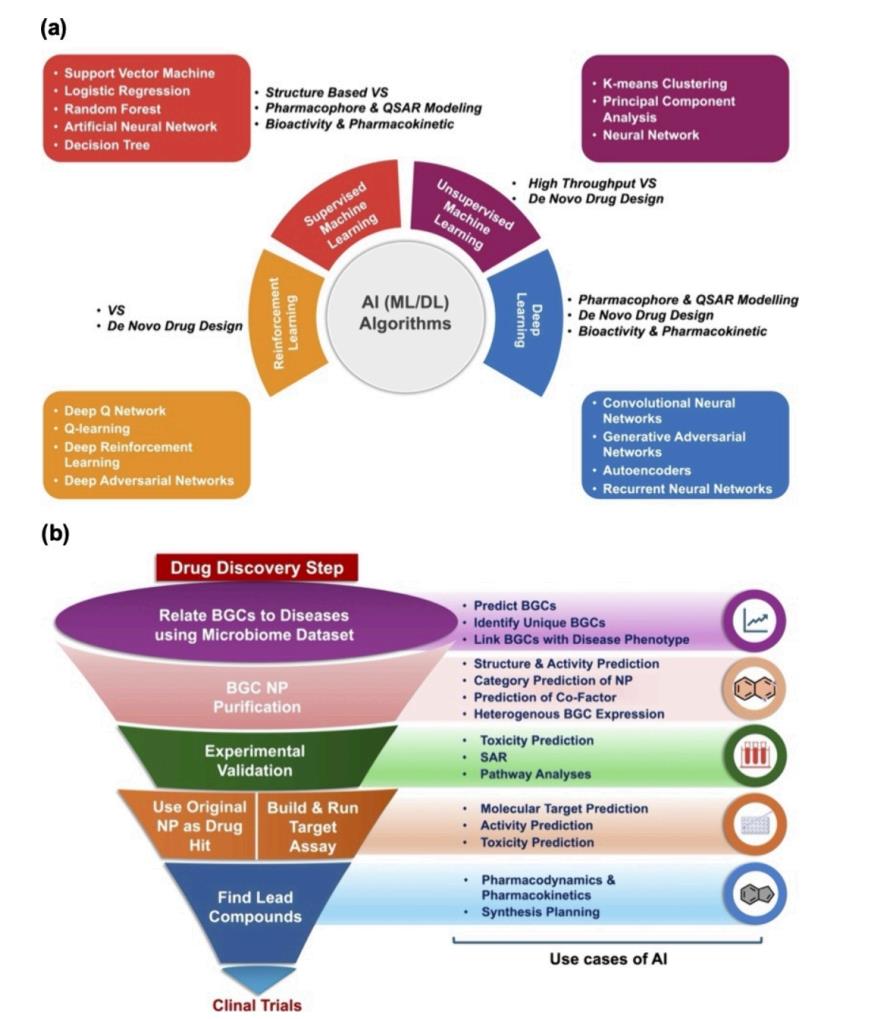
Fig.2 AI-driven drug discovery approaches.1
What the Near Future of NP Drug Discovery Likely Looks Like
Deeper Generative/Retrosynthetic Coupling. Generative models will soon co-optimize potency, selectivity, and synthesizability in a single loop, with reinforcement learning steering exploration toward NP-like chemical spaces while maintaining synthetic feasibility.
NP-Aware Design Spaces. AI models fine-tuned on NP fragments and biosynthetic logic will yield “pseudo-natural” chemotypes with NP-like topology but improved developability, accelerating hit-to-lead transitions with fewer synthetic obstacles.
Multimodal Knowledge Graphs at Scale. Integrating spectral (NMR/MS), genomic (BGC), and bioassay data will empower graph-based rankers and retrieval-augmented models to answer complex design questions and propose data-backed molecular hypotheses.
Standardized Pipelines and Benchmarks. As open-source retrosynthesis and virtual screening tools mature, NP-focused teams will converge on reproducible workflows, promoting transparency and method standardization across AI-driven discovery pipelines.
Practical Playbook: Building an AI-Forward NP Drug Discovery Pipeline
-
Curate Once, Benefit Everywhere. Normalize NP libraries with computable chemical classes and unified identifiers—foundational steps that enhance every downstream application from QSAR to generative modeling.
-
Fuse Upstream Signals. Combine genomic, metabolomic, and dereplication analytics to prioritize isolation efforts toward biosynthetically plausible novelty with disease-relevant profiles.
-
Use NP-Likeness and Synthesizability Filters. Before synthesis, employ NP-likeness scorers and retrosynthesis planners to identify makeable, structurally credible NP-inspired compounds.
-
Go Generative—Carefully. Fine-tune transformer or VAE models on NP-specific data while constraining with property and routeability objectives. Collaborate closely with chemists for interpretable, iterative SAR optimization.
-
Make Decisions Explainable. Apply XAI to visualize substructure relevance and quantify uncertainty—critical for confidence when extrapolating beyond known NP chemical space.
Bottom Line: From Search to Design
AI has reshaped NP drug discovery by linking biosynthesis and pharmacology through shared, computable frameworks. When organizations invest in data curation, adopt NP-aware generative models, and integrate synthesis feasibility checks, “natural-product-inspired design” evolves from concept to a reproducible, industrially scalable reality.
Services We Recommend (Quick List)
-
AI-Antibody Solution – de novo sequence generation, developability prediction, and rapid property optimization.
-
AI Drug Discovery Services – NP-aware virtual screening, ML/QSAR, and generative design tied to scalable validation.
-
AI One-Stop Antibody Discovery Platform – closed-loop design–build–test–learn workflows with explainable AI integration.
-
AI One-Stop Drug Discovery Platform – end-to-end small-molecule discovery integrating knowledge graphs, generative chemistry, and retrosynthesis intelligence.
Reference:
1.Gangwal, Amit, and Antonio Lavecchia. “Artificial intelligence in natural product drug discovery: current applications and future perspectives.” Journal of medicinal chemistry 68.4 (2025): 3948-3969. Distributed under Open Access license CC BY 4.0, without modification. https://doi.org/10.1021/acs.jmedchem.4c01257
2. Merk, Daniel, et al. “Tuning artificial intelligence on the de novo design of natural-product-inspired retinoid X receptor modulators.” Communications Chemistry 1.1 (2018): 68. https://doi.org/10.1038/s42004-018-0068-1